

MT Ferrule Market Summary
MT ferrule is a ferrule component commonly used in connectors, fiber optic equipment and other high-precision electronic equipment, especially in the field of fiber optic communications. It is the core part of the plug or connector, usually used for optical fiber connection and signal transmission. MT ferrule consists of multiple fiber channels, each channel can accommodate one optical fiber, the number of channels is generally 12, 24 or more, and can handle multiple fiber optic signals at the same time. MT ferrule is usually composed of a metal shell and a precision plastic base. The role of the shell is to provide protection for the internal optical fiber and ensure the stability and anti-interference ability of the connector.
According to the new market research report “Global MT Ferrule Market Report 2026-2032”, published by QYResearch, the global MT Ferrule market size is projected to reach USD 0.46 billion by 2032, at a CAGR of 7.9% during the forecast period.
Figure00001. Global MT Ferrule Market Size (US$ Million), 2021-2032
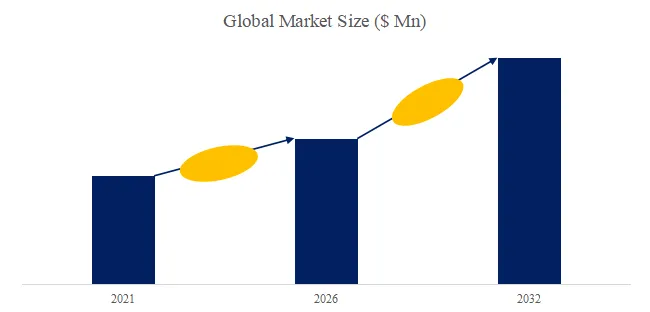
Above data is based on report from QYResearch: Global MT Ferrule Market Report 2026-2032 (published in 2026). If you need the latest data, plaese contact QYResearch.
Figure00002. Global MT Ferrule Top 14 Players Ranking and Market Share (Ranking is based on the revenue of 2025, continually updated)
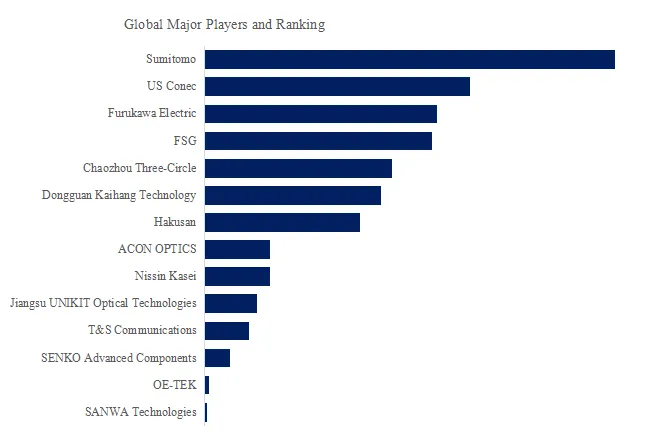
Above data is based on report from QYResearch: Global MT Ferrule Market Report 2026-2032 (published in 2026). If you need the latest data, plaese contact QYResearch.
According to QYResearch Top Players Research Center, the global key manufacturers of MT Ferrule include Sumitomo, US Conec, Furukawa Electric, FSG, Chaozhou Three-Circle, Dongguan Kaihang Technology, Hakusan, ACON OPTICS, Nissin Kasei, Jiangsu UNIKIT Optical Technologies, etc. In 2025, the global top five players had a share approximately 55.0% in terms of revenue.
Figure00003.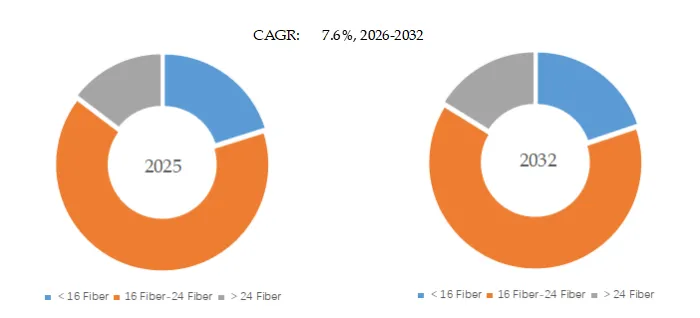 MT Ferrule, Global Market Size, Split by Product Segment
MT Ferrule, Global Market Size, Split by Product Segment
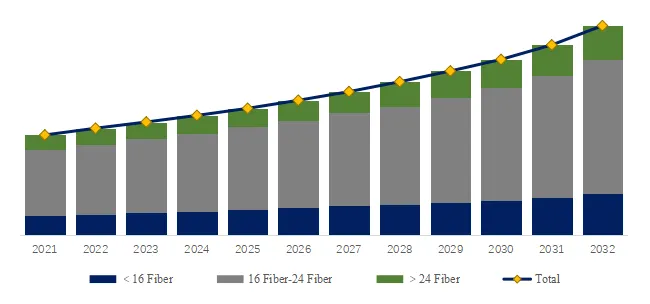
Based on or includes research from QYResearch: Global MT Ferrule Market Report 2026-2032.
In terms of product type, 16 Fiber-24 Fiber is the largest segment, hold a share of 65.2%,
Market Drivers:
Rapid Development of Data Centers and Cloud Computing
With the explosive growth of cloud computing, big data, and AI applications, the construction of hyperscale data centers is accelerating, significantly increasing the demand for high-speed, high-density optical connections. MT ferrules, as the core component of MPO/MTP optical connectors, enable parallel transmission of multi-core optical fibers and are a key fundamental component for high-bandwidth cabling in data centers, thus driving continuous industry growth.
5G and Future Communication Network Construction
5G base stations and subsequent communication networks place higher demands on high-speed optical fiber connections, especially in fronthaul, midhaul, and backhaul networks, requiring numerous high-density optical connection solutions. MT ferrules, with their advantages of high multi-core alignment accuracy and high transmission efficiency, have become an important supporting product for communication network upgrades.
Upgrades in Optical Modules and High-Speed Transmission Technology
The gradual commercialization of high-speed optical modules such as 400G/800G has continuously increased the requirements for connector accuracy, insertion loss, and stability. As a core precision alignment structure, the processing accuracy and consistency of MT ferrules directly affect the performance of optical modules. Therefore, the continuous upgrade of optical communication rates is driving the advancement of MT ferrule technology and the growth of market demand.
Artificial Intelligence and Computing Infrastructure Construction
AI training and inference have extremely high demands for computing power and data transmission bandwidth, driving the construction of GPU clusters and high-speed interconnect networks. High-density optical connections have become a critical infrastructure, and MT ferrules are indispensable in parallel optical interconnects, leading to a rapid increase in their demand with the construction of AI computing centers.
Restraint:
High-precision manufacturing technology has high barriers to entry. MT ferrules are micron-level precision optical communication devices, requiring extremely high precision in hole positioning, end-face flatness, and coaxiality. This necessitates strong reliance on ceramic material processing, mold design, and micro-hole machining technologies. High-end product yield control is difficult, and small and medium-sized enterprises struggle to overcome core process barriers, limiting the overall supply capacity of the industry.
Upstream reliance on key equipment and materials. MT ferrule production involves high-precision drilling equipment, grinding equipment, and high-performance ceramic powder materials. Some core equipment and materials still rely on imports. Supply chain fluctuations or technological limitations will affect production stability and cost control, constraining industry development.
Downstream demand exhibits significant cyclical fluctuations. MT ferrules are mainly used in data centers and communication networks, industries heavily influenced by cloud vendors’ capital expenditures (Capex) and operator investment cycles. When data center construction slows or communication investment cycles decline, MT ferrule demand may experience periodic fluctuations.
Intensified price competition within the industry. As the market expands and new entrants increase, price competition has emerged in some low-to-mid-end products, squeezing profit margins for companies. Especially in product sectors with a high degree of standardization, companies are prone to falling into a competitive landscape of “trading volume for price,” which is not conducive to the long-term healthy development of the industry.
Opportunity:
The accelerated construction of AI and computing centers is driving the explosive growth in demand for generative AI, large-scale model training, and high-performance computing. This is propelling the construction of ultra-large-scale computing centers and placing higher demands on high-bandwidth, low-latency data transmission. MT ferrules, as core components of MPO/MTP multi-core connections, are indispensable in parallel optical interconnects and directly benefit from the continuous expansion of AI computing infrastructure.
Data centers are evolving towards higher speeds and densities. The increasing prevalence of 400G, 800G, and even higher-speed optical modules is driving the upgrade of data center cabling towards higher density and lower loss. MT ferrules enable high-precision alignment of multi-core optical fibers and are a key component for achieving high-density connections; their demand will continue to grow with the upgrade of data center network architecture.
The upgrading of 5G/6G and fiber optic communication networks is also driving the expansion of MT ferrule applications in communication infrastructure. The deepening construction of 5G networks and the future evolution of 6G technology are placing higher capacity and lower latency requirements on fronthaul, midhaul, and backhaul networks. The development of fiber optic networks towards greater bandwidth and higher connection density is expanding the application scale of MT ferrules in communication infrastructure. The development of optical modules and silicon photonics technology drives demand for related components. The rapid development of silicon photonics technology and high-speed optical modules has enabled optical interconnects to evolve towards higher integration and lower power consumption, but it also places higher demands on optical connection accuracy. MT ferrules, as a key passive alignment structure, will continue to play a vital role in high-end optical modules and optical interconnect solutions, enhancing product added value.
About The Authors
| Ziyi Fan | ||
| Lead Author | ||
| Consumer Goods,
Equipment & Parts, Packaging, etc. |
About QYResearch
QYResearch founded in California, USA in 2007.It is a leading global market research and consulting company. With over 19 years’ experience and professional research team in various cities over the world QY Research focuses on management consulting, database and seminar services, IPO consulting (data is widely cited in prospectuses, annual reports and presentations), industry chain research and customized research to help our clients in providing non-linear revenue model and make them successful. We are globally recognized for our expansive portfolio of services, good corporate citizenship, and our strong commitment to sustainability. Up to now, we have cooperated with more than 60,000 clients across five continents. Let’s work closely with you and build a bold and better future.
QYResearch is a world-renowned large-scale consulting company. The industry covers various high-tech industry chain market segments, spanning the semiconductor industry chain (semiconductor equipment and parts, semiconductor materials, ICs, Foundry, packaging and testing, discrete devices, sensors, optoelectronic devices), photovoltaic industry chain (equipment, cells, modules, auxiliary material brackets, inverters, power station terminals), new energy automobile industry chain (batteries and materials, auto parts, batteries, motors, electronic control, automotive semiconductors, etc.), communication industry chain (communication system equipment, terminal equipment, electronic components, RF front-end, optical modules, 4G/5G/6G, broadband, IoT, digital economy, AI), advanced materials industry Chain (metal materials, polymer materials, ceramic materials, nano materials, etc.), machinery manufacturing industry chain (CNC machine tools, construction machinery, electrical machinery, 3C automation, industrial robots, lasers, industrial control, drones), food, beverages and pharmaceuticals, medical equipment, agriculture, etc.
About Us:
QYResearch founded in California, USA in 2007, which is a leading global market research and consulting company. Our primary business include market research reports, custom reports, commissioned research, IPO consultancy, business plans, etc. With over 18 years of experience and a dedicated research team, we are well placed to provide useful information and data for your business, and we have established offices in 7 countries (include United States, Germany, Switzerland, Japan, Korea, China and India) and business partners in over 30 countries. We have provided industrial information services to more than 60,000 companies in over the world.
Contact Us:
If you have any queries regarding this report or if you would like further information, please contact us:
QY Research Inc.
Add: 17890 Castleton Street Suite 369 City of Industry CA 91748 United States
EN: https://www.qyresearch.com
Email: global@qyresearch.com
Tel: 001-626-842-1666(US)
JP: https://www.qyresearch.co.jp







